Introduction to Metadata and Ontologies
Everything You Always Wanted to Know About Metadata and Ontologies (But Were Afraid to Ask)
About Metadata
What are metadata?
Metadata are contextual data about your experimental data. Metadata are the who, what, when, where, and why of these data. Metadata put these data into context. In microbiome research, metadata include information about the sample: when it was collected, where it was collected from, what kind of sample it is, and what were the properties of the environment or experimental condition from which the sample was taken. Information about sample processing is also metadata: methods used to extract and purify molecules (e.g., DNA) from the sample, type of DNA sequencing or other ’omics analyses done, and where the raw experimental data are located.
What kinds of metadata are there?
For a microbiome study, metadata exist at multiple stages along the path from samples to analysis. For example, contextual information about when and where the sample was collected could include descriptors like date, time, and geospatial coordinates. Processing the sample in the lab for analysis requires different protocols and instrumentation. Once these data become electronic and are analyzed, the results should be accompanied by the versioned software used.
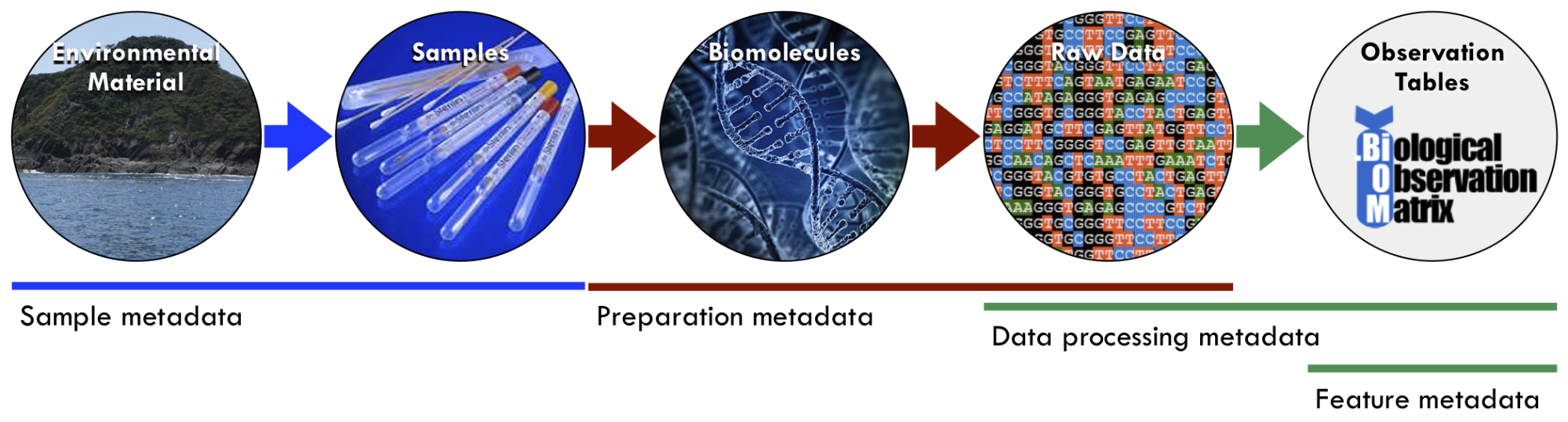
Figure 1. Examples of different types of metadata along the workflow from environmental samples to data and analysis tables. Submitting data to central repositories typically requires sample and preparation metadata. Data processing and feature metadata are generated by the repository or analysis software. Credit: Luke Thompson, PhD (National Oceanic and Atmospheric Administration).
- Sample metadata – Information about the primary sample: when it was collected (e.g., date and time), where it was collected from (e.g., latitude, longitude, elevation/depth, site name, country, etc.), what kind of sample it was (e.g., soil, seawater, feces), and the properties of the environment during collection (e.g., temperature, salinity, pH) or experimental condition (e.g., experimental or control, disease state) from which the sample was taken. Sample metadata are not dependent on how the sample was processed; that is called preparation metadata (see next item).
- Preparation metadata– Information about preparation of the primary sample. A primary sample could be split (aliquoted) and processed through multiple preparation methods; therefore, there could be multiple sets of preparation metadata for a single set of samples. For DNA sequencing of a microbiome sample, preparation metadata include the type of DNA extraction method, conditions used for sequencing (e.g., primers, library kits, sequencing instrumentation and parameters), and where the raw sequence data sets are accessible.
- Data processing metadata – Data about the properties and downstream processing of the raw experimental data from each sample, including software parameters and version numbers. For example, if DNA sequences were generated, this could include the sequence properties (e.g., sequence lengths, sequences per sample, and total base pairs), quality control and filtering (e.g., adapter trimming, quality trimming and filtering, dereplicating, and chimera removal), assembly parameters (e.g., assembly tool, binning tool, and finishing strategy), gene annotation (e.g., gene calling tool and annotation database), and other processing parameters.
- Feature metadata – Data about features detected in the samples, rather than about the samples themselves. For example, if amplicon sequencing was done, feature metadata might include information (e.g., taxonomy, reference sequences, and sequence identifiers) about the OTUs or ASVs generated in the OTU-picking or denoising algorithm. If metabolomics analysis was done, feature metadata might include information (e.g., MS2 fragments produced or candidates for identification) about the metabolites detected.
Why are metadata important?
Hypotheses are driven by and dependent on metadata; in other words, metadata are critically important for answering scientific questions. In the context of microbiome samples, the identification of a bacterial community and what it was doing is not very meaningful without descriptive variables that enable comparisons (e.g., among groups, with other covariates, etc.). For example, if you were presented with a box of canned foods without labels, you may be able to group similar items based on their contents, but you would be limited from asking more meaningful questions (e.g., salt concentration differences between two manufacturers of the same kind of soup, or degradation of nutrients based on shelf life). Simply put, you cannot analyze your data if you don’t know the properties of your samples, where they come from, and how they relate to each other.
Metadata are also critical for the sharing and reuse of scientific data. For other researchers to use your data with other data sets, they need to know about your samples and data. Basically, they need your metadata in a format compatible with their metadata. The principles of data sharing are encapsulated by the FAIR Data Principles, which states that data should be Findable, Accessible, Interoperable, and Reusable. The criteria of the FAIR principles are as follows:
- Findable – Metadata should be richly described, include the identifier of the data they describe, be assigned a globally unique and persistent identifier, and be indexed in a searchable resource.
- Accessible – Metadata should be retrievable by an identifier using a standardized communications protocol, which is open, free, and universally implementable, and the metadata should be accessible even when the data are no longer available.
- Interoperable – Metadata should use a formal, accessible, shared, and broadly applicable language for knowledge representation, using vocabularies that follow FAIR principles, and include qualified references to other metadata and/or data.
- Reusable – Metadata are richly described with a plurality of accurate and relevant attributes, are released with a clear and accessible data usage license, are associated with detailed provenance, and meet domain-relevant community standards.
Why should metadata be standardized?
Despite the critical nature of metadata, metadata collection is often poorly standardized and error-prone. Tabular formats (such as Microsoft Excel) continue to be popular options for metadata collection and record-keeping, yet freeform text entry without validation is prone to errors (e.g., misspellings, incorrect data, missing data, and inconsistent values). These issues can emerge within a single study and researcher, and are even more likely across multiple researchers and/or studies.
In short, standardized metadata encourage data reuse. For example, with standardized metadata, experimental results from different labs can be grouped together for combined studies with a scope that can extend beyond what can be done from a single lab. It also lays the foundation for researchers to quickly find previous experiments of interest to them.
Why should metadata be machine readable?
In order for all of these metadata to be discoverable by machines as well as by people, each component must be described in a uniform way. For example, a person can easily distinguish similarities and differences between the words: apple, red delicious, granny smith, banana, and fruit (below), but without a structured relationship, a computer cannot.
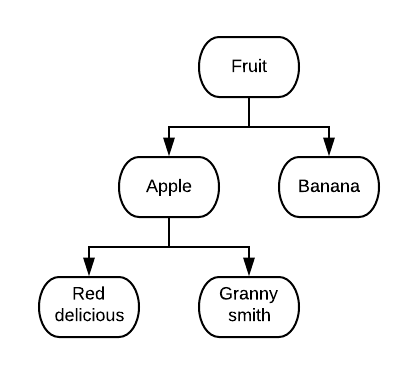 Structured relationships between terms to describe data are called ontologies. One of the most basic forms of an ontology is a hierarchical classification, which uses an “is a” relationship between terms. Sample metadata can also be structured to use ontologies. The structured relationships provided by ontologies make our metadata more machine readable.
Structured relationships between terms to describe data are called ontologies. One of the most basic forms of an ontology is a hierarchical classification, which uses an “is a” relationship between terms. Sample metadata can also be structured to use ontologies. The structured relationships provided by ontologies make our metadata more machine readable.
About Ontologies
What are ontologies?
From Ontotext:
An ontology is a formal description of knowledge as a set of concepts within a domain and the relationships that hold between them. To enable such a description, we need to formally specify components such as individuals (instances of objects), classes, attributes and relations as well as restrictions, rules and axioms. As a result, ontologies do not only introduce a shareable and reusable knowledge representation but can also add new knowledge about the domain.
The relationships between concepts can occur within a single ontology, or be used to link knowledge across ontologies.
For example, let’s talk about how x links to y via z. We use multiple ontologies to describe environmental chemical measurements, e.g., nitrate in water. The Environment Ontology (ENVO) provides a term for the environment of liquid water; the Chemical Entities of Biological Interest ontology (CHEBI) provides a term for nitrate; and the Phenotype and Trait Ontology (PATO) provides a term for the concentration of nitrate in water. Putting this all together, and using machine readable formatting, here it is in English and then coded:
A sample of liquid water (ENVO) is sent for processing to determine the concentration of (PATO) nitrate (CHEBI).
PATO:’concentration of’ and (PATO:’inheres in’ some (CHEBI:nitrate and (‘part of’ some ENVO:’liquid water’))) By using precise language to describe these relationships, both humans and machines are able to reason on the same data.
Ontologies are also adaptable. As knowledge in each domain expands and new relationships are formed, the ontologies grow alongside it. This flexibility also allows for cross-referencing to other controlled vocabularies or ontologies. For example, an ontology related to food could ensure that fries (in the American English) and chips (in British English) are machine-readable as the same food (defined as thin strips of deep-fried potato).
Why are ontologies important?
Researchers need specific data in order to answer specific questions. Standardized metadata are important for enabling search based on matches to specific terms across data sets; however, sometimes there is a need for more complex queries. Ontologies are helpful when organizing data, but they also enable powerful queries that make use of the terms and the relationships between terms. Using the example above, a simple query could discover the concentration of nitrogen in liquid water, or more specific water types, such as lakes, rivers, or oceans.
What are some examples of ontologies?
Domain ontologies exist for many different areas of study:
- Environment (ENVO)
- Chemistry (CHEBI)
- Genes in organisms (GO)
- Phenotypes and traits (PATO)
- Anatomy (UBERON)
Further Resources
Metadata guides and tools
These resources from our partner organizations provide further guidance for creating metadata:
- Open Biological and Biomedical Ontology (OBO) Foundry
- Minimum Information about any (x) Sequence (MIxS) Standard from Genomic Standards Consortium (GSC)
- Earth Microbiome Project (EMP) Metadata Guide
- Quick and Intuitive Interactive Metadata Portal (QIIMP)
Tips for formatting metadata for maximum utility
Our experience working with diverse sample sets has taught us some principles for formatting metadata:
- Simple tabular format – Metadata should be formatted as a simple table with one row at the top for column headers and one column at the left for sample names. Sticking to lowercase letters and underscores in column headers makes it easier when using software (e.g., Python, R) to analyze the data. Resist the urge to add cells with notes on the margins; instead try to find a way to have any information you want conveyed within the strict structure of rows and columns, where each row is a sample and each column is a property of the samples. See the MIxS templates as a starting point for entering metadata.
- Text only, no colors – Colors and highlighting aren’t good to use in metadata, because they’re not machine-readable. If you load the table into Python or R, or convert to csv or tsv, you won’t see the colors or highlighting. Any information you want to be kept should be written out in one of the columns.
- Add your own columns that group the samples into experimentally meaningful groups – Add columns that might be useful in analyzing these data. Here you can think about the samples and what distinguishes them from each other. For example, let’s say you have samples from a harmful algal bloom in a lake. Were some samples collected at day, others at night? Some deep, some shallow? Some inside the bloom, some outside? Anything like this could be summarized in a column, such as for example, “proximity_to_bloom” with values “inside” or “outside”. Or maybe include a column “control_status” with values “negative control”, “mock community”, “single culture”, or “biological sample”. All of this will make analyzing and graphing the data easier.
- One sample metadata file, multiple other metadata files – As noted in the section about the different kinds of metadata, often we record sample metadata separately from prep metadata. This is useful because sometimes a single biological sample may have multiple filters (extra rows in the prep metadata sheet) and multiple preps (extra sheets of prep metadata). The sample and prep metadata can always be merged later using Python or R.
Contributors:
Luke Thompson, National Oceanic and Atmospheric Administration
Kai Blumberg, University of Arizona
Danielle Christianson, Lawrence Berkeley National Laboratory
Jose Pablo Dundore-Arias, California State University, Monterey Bay
Bin Hu, Los Alamos National Laboratory
Ruth Timme, U.S. Food and Drug Administration
Pajau Vangay, Lawrence Berkeley National Laboratory
Elisha Wood-Charlson, Lawrence Berkeley National Laboratory
To reference this document in a publication, please use the following citation:
Thompson, Luke, Vangay, Pajau, Blumberg, Kai, Christianson, Danielle, Dundore-Arias, Jose Pablo, Hu, Bin, Timme, Ruth, and Wood-Charlson, Elisha M. Introduction to Metadata and Ontologies: Everything You Always Wanted to Know About Metadata and Ontologies (But Were Afraid to Ask). United States: N. p., 2020. Web. doi:10.25979/1607365.